
VALPROSID
Estás aquí
Contáctanos

Forma farmacéutica: Solución
Cada ampolleta contiene:
Valproato de sodio ................... 400 mg
Vehículo cbp............................. 4 mL
VALPROSID® está indicado en el manejo agudo de la epilepsia, tanto parcial como generalizada.
Como alternativa intravenosa en pacientes en quienes la administración por vía oral, de otros productos de valproato, no es factible temporalmente, en las siguientes condiciones:
Como monoterapia o tratamiento adjunto en pacientes con crisis parciales complejas que ocurren aisladas o en asociación con otros tipos de crisis.
También está indicado como monoterapia o terapia complementaria para el tratamiento de las crisis de ausencia simple y compleja, y de manera complementaria en pacientes con múltiples tipos de crisis que incluyen crisis de ausencias.
El valproato de sodio (solución inyectable) no debe administrarse a pacientes con enfermedad o disfunción hepáticas significativa (ver Precauciones generales).
El valproato de sodio está contraindicado en pacientes con enfermedad de Alpers o Alpers-Huttenlocher (ver Precauciones generales). Niños menores de 2 años con sospecha de tener un trastorno relacionado con POLG, trastornos mitocondriales causados por mutaciones en la polimerasa y ADN Mitocondrial.
El valproato de sodio está contraindicado en pacientes con hipersensibilidad conocida al medicamento.
El valproato de sodio está contraindicado en pacientes con trastorno en el ciclo de la urea (ver Precauciones generales).
Pacientes con Porfiria, Embarazo y Lactancia.
Los eventos adversos que induce la inyección de valproato de sodio incluyen a los observados con formas orales del mismo. Lo siguiente describe la experiencia específicamente con valproato de sodio inyectable. La forma inyectable, por lo general, ha sido bien tolerada en ensayos clínicos realizados en voluntarios adultos masculinos (111) y en pacientes epilépticos (352), en dosis de 125 a 6000 mg (dosis total/día).
Un total de 2% de pacientes descontinuaron el tratamiento con valproato de sodio inyectable debido a eventos adversos, siendo los más comunes (2 casos cada uno) náusea/vómito y amilasa elevada. Otros, que llevaron a la suspensión del uso del fármaco fueron: alucinaciones, neumonía, cefalea, reacción en el sitio de inyección y marcha anormal. El mareo y el dolor en el sitio de inyección fueron observados con mayor frecuencia con una velocidad de infusión de 100 mg/min, que a velocidades de hasta 33 mg/min. A velocidad de 200 mg/min, el mareo y la alteración del gusto ocurrieron con mayor frecuencia que a 100 mg/min. La velocidad máxima de infusión estudiada fue de 200 mg/min.
Los eventos adversos informados por al menos 0.5% de todos los sujetos/pacientes en los ensayos clínicos con valproato de sodio inyectable se muestran, en resumen, en la tabla 1.
Tabla 1. Eventos adversos reportados durante estudios con valproato de sodio inyectable
|
Sistema corporal/evento |
n = 463 |
|
Cuerpo como un todo |
|
|
Dolor torácico |
1.7% |
|
Cefalea |
4.3% |
|
Inflamación en el sitio de inyección |
0.6% |
|
Dolor en el sitio de inyección |
2.6% |
|
Reacción en el sitio de inyección |
2.4% |
|
Dolor (inespecífico) |
1.3% |
|
Cardiovascular |
|
|
Vasodilatación |
0.9% |
|
Dermatológico |
|
|
Sudoración |
0.9% |
|
Sistema digestivo |
|
|
Dolor abdominal |
1.1% |
|
Diarrea |
0.9% |
|
Náusea |
3.2% |
|
Vómito |
1.3% |
|
Sistema nervioso |
|
|
Mareo |
5.2% |
|
Euforia |
0.9% |
|
Hiperestesia |
0.6% |
|
Nerviosismo |
0.9% |
|
Parestesia |
0.9% |
|
Somnolencia |
1.7% |
|
Temblor |
0.6% |
|
Respiratorio |
|
|
Faringitis |
0.6% |
|
Sentidos especiales |
|
|
Alteración del gusto |
1.9% |
Epilepsia:
Crisis parciales complejas: Con base en ensayos controlados con placebo del tratamiento adjunto para el tratamiento de las crisis parciales complejas (CPC), por lo general, el divalproato de sodio fue bien tolerado, con la mayoría de eventos adversos calificados de severidad leve a moderada. La intolerancia fue la principal razón para la descontinuación en los pacientes tratados con divalproato de sodio (6%) comparado con los pacientes tratados con placebo (1%).
La tabla 2 enlista los eventos adversos que fueron reportados por ≥ 5% de los pacientes tratados con divalproato de sodio, para los cuales la incidencia fue mayor que en el grupo placebo. Ya que los pacientes también fueron tratados con otros fármacos antiepilépticos, no es posible, en la mayoría de los casos, determinar si los siguientes eventos adversos pueden ser atribuidos a valproato de sodio solamente o a la combinación de valproato con otros fármacos antiepilépticos.
Tabla 2. Eventos adversos informados por ≥ 5% de pacientes tratados con valproato semisódico durante ensayos controlados acerca del tratamiento conjunto para convulsiones parciales complejas
|
Sistemas corporales/eventos |
DVP* (%) (n = 77) |
Placebo (%) (n = 70) |
|
Cuerpo total |
||
|
Cefalea |
31 |
21 |
|
Astenia |
27 |
7 |
|
Fiebre |
6 |
4 |
|
Sistema gastrointestinal |
||
|
Náusea |
48 |
14 |
|
Vómito |
27 |
7 |
|
Dolor abdominal |
23 |
6 |
|
Diarrea |
13 |
6 |
|
Anorexia |
12 |
0 |
|
Dispepsia |
8 |
4 |
|
Constipación |
5 |
1 |
|
Sistema nervioso |
||
|
Somnolencia |
27 |
11 |
|
Temblor |
25 |
6 |
|
Mareo |
25 |
13 |
|
Diplopía |
16 |
9 |
|
Ambliopía/visión borrosa |
12 |
9 |
|
Ataxia |
8 |
1 |
|
Nistagmus |
8 |
1 |
|
Labilidad emocional |
6 |
4 |
|
Pensamiento anormal |
6 |
0 |
|
Amnesia |
5 |
1 |
|
Sistema respiratorio |
||
|
Síndrome gripal |
12 |
9 |
|
Infección |
12 |
6 |
|
Bronquitis |
5 |
1 |
|
Rinitis |
5 |
4 |
|
Otros |
||
|
Alopecia |
6 |
1 |
|
Pérdida de peso |
6 |
0 |
|
*DVP = Divalproato de sodio. |
|
|
En la tabla 3 se enlistan eventos adversos que surgen durante el tratamiento y que fueron informados por ≥ 5% de pacientes en el grupo con la dosis más alta de divalproato de sodio y en quienes la incidencia fue mayor para el grupo con la dosis más baja, en ensayos como monoterapia de convulsiones parciales complejas. Puesto que, durante la primera parte del ensayo, los pacientes habían sido titulados en retirada de otro fármaco antiepiléptico, no es posible en muchos casos determinar en qué casos los siguientes eventos adversos pueden ser atribuidos al divalproato de sodio solo, o a la combinación del valproato semisódico y otros fármacos antiepilépticos.
Tabla 3. Eventos adversos informados en ≥ 5% de los pacientes en el grupo con dosis alta de valproato semisódico en monoterapia en pacientes con crisis parciales complejas1
|
Sistema corporal/ |
Dosis alta (%) (n = 131) |
Dosis baja (%) (n = 134) |
|
Cuerpo como un todo |
||
|
Astenia |
21 |
10 |
|
Sistema digestivo |
||
|
Náusea |
34 |
26 |
|
Diarrea |
23 |
19 |
|
Vómito |
23 |
15 |
|
Dolor abdominal |
12 |
9 |
|
Anorexia |
11 |
4 |
|
Dispepsia |
11 |
10 |
|
Sistema hemo/linfático |
||
|
Trombocitopenia |
24 |
1 |
|
Equimosis |
5 |
4 |
|
Metabólico/nutricional |
||
|
Aumento de peso |
9 |
4 |
|
Edema periférico |
8 |
3 |
|
Sistema nervioso |
||
|
Temblor |
57 |
19 |
|
Somnolencia |
30 |
18 |
|
Mareo |
18 |
13 |
|
Insomnio |
15 |
9 |
|
Nerviosismo |
11 |
7 |
|
Amnesia |
7 |
4 |
|
Nistagmo |
7 |
1 |
|
Depresión |
5 |
4 |
|
Sistema respiratorio |
||
|
Infección |
20 |
13 |
|
Faringitis |
8 |
2 |
|
Disnea |
5 |
1 |
|
Piel y anexos |
||
|
Alopecia |
24 |
13 |
|
Sentidos especiales |
||
|
Visión borrosa |
8 |
4 |
|
Tinnitus |
7 |
1 |
1Cefalea fue el único evento adverso que ocurrió en ≥ 5% de pacientes en el grupo con dosis alta y una incidencia igual o mayor en el grupo con dosis baja.
Los siguientes eventos adversos adicionales fueron informados en proporción > 1% y < 5% en 358 pacientes tratados con divalproato de sodio, en los ensayos controlados, por convulsiones parciales complejas.
Cuerpo total: Dolor de espalda, dolor torácico y malestar.
Cardiovascular: Taquicardia, hipertensión y palpitaciones.
Digestivo: Hiperfagia, flatulencia, hematemesis, eructos, pancreatitis y abscesos periodontales.
Hemo/linfático: Petequias.
Metabólico/nutricional: Aumento de TGO y TGP.
Musculoesquelético: Mialgia, contracciones, artralgias, calambres en las piernas y miastenia.
Sistema nervioso: Ansiedad, confusión, trastornos del lenguaje, marcha anormal, parestesias, hipertonía, incoordinación, sueños anormales y trastornos de personalidad.
Respiratorio: Sinusitis, aumento de tos, neumonía y epistaxis.
Piel y anexos: Salpullido, prurito y piel seca.
Sentidos especiales: Alteración del gusto, visión borrosa, trastornos en la audición, sordera y otitis media.
Urogenital: Incontinencia urinaria, vaginitis, dismenorrea, amenorrea y frecuencia urinaria.
Otras poblaciones de pacientes: A continuación, se enumeran, por sistema y aparato del organismo, los eventos adversos que se han reportado con todas las formas farmacéuticas de valproato de estudios de epilepsia, reportes espontáneos y otras fuentes.
Gastrointestinales: Los efectos reportados más comúnmente en el inicio del tratamiento son náusea, vómito e indigestión. Estos efectos generalmente son transitorios y rara vez es necesario suspender el tratamiento. Se han reportado diarrea, cólicos y estreñimiento. También se ha reportado anorexia con cierta pérdida de peso y aumento de apetito con aumento de peso. La administración de valproato semisódico con cubierta entérica puede resultar en la reducción de efectos secundarios gastrointestinales en algunos pacientes.
Efectos en el SNC: Se han observado efectos sedantes en pacientes que reciben valproato solo, pero se presentan con más frecuencia en pacientes que reciben tratamiento combinado. La sedación por lo regular disminuye al reducir el otro medicamento antiepiléptico. Se reportaron temblor (puede relacionarse con la dosis), alucinaciones, ataxia, cefalea, nistagmus, diplopía, asterixis, “manchas delante de los ojos”, disartria, mareo, confusión, hipoestesia, vértigo, falta de coordinación y parkinsonismo con el uso de valproato. Se han presentado casos contados de coma en pacientes que reciben valproato solo o en combinación con fenobarbital. En contados casos se ha desarrollado encefalopatía con o sin fiebre poco después de la introducción de monoterapia con valproato sin evidencia de disfunción hepática o niveles plasmáticos de valproato inadecuadamente altos. A pesar de que se ha definido una recuperación después de retirar el fármaco, han habido muertes entre los pacientes con encefalopatía hiperamonémica, en particular en pacientes con trastornos subyacentes del ciclo de la urea (ver Precauciones generales).
Diversos reportes han mencionado demencia reversible y pseudoatrofia reversible en asociación con el tratamiento con valproato.
En un estudio de 26 meses, controlado con placebo, llevado a cabo por los Institutos Nacionales de Salud en Enfermedad de Alzheimer (Aging-Alzheimer’s Disease Cooperative Study [NIA-ADCS]) se incluyeron 313 pacientes con enfermedad leve a moderada de Alzheimer para evaluar el beneficio de una terapia a dosis baja de valproato (10 mg/kg/d) en el retraso de la aparición de agitación y/o psicosis, y la atenuación de la progresión clínica de la enfermedad. Los resultados del estudio no demostraron beneficio del tratamiento con valproato. En el grupo de valproato, las calificaciones de la prueba Mini-Mental Status Examination (MMSE) mostraron una declinación más rápida en el mes 12 (placebo: -2.3 ± 4.3; valproato: -3.9 ± 4.0) pero no hubo cambios en la prueba ADAS-cog u otras pruebas de conducta o funcionales (actividades de la vida diaria) medidas en los meses 12 o 24. Se desconoce el significado clínico de estos hallazgos.
Dermatológicos: Pérdida de cabello transitoria, erupción cutánea, fotosensibilidad, prurito generalizado, eritema multiforme y síndrome de Stevens-Johnson. Se han reportado contados casos de necrólisis epidérmica tóxica incluyendo un caso mortal en un lactante de seis meses de edad que tomaba valproato y otros medicamentos concomitantes. Se reportó otro caso más de necrólisis epidérmica tóxica que causó la muerte a un paciente de 35 años con SIDA que tomaba diversos medicamentos concomitantes y con antecedentes de múltiples reacciones cutáneas a fármacos. Se han reportado reacciones cutáneas serias con la administración concomitante de lamotrigina y valproato.
Psiquiátricos: Alteración emocional, depresión, psicosis, agresión, hiperactividad, hostilidad y deterioro de la conducta.
Musculoesqueléticos:
Debilidad: Se han recibido reportes de disminución de masa ósea, derivando posiblemente en osteoporosis y osteopenia, durante el tratamiento a largo plazo con medicamentos anticonvulsivos, incluyendo valproato. Algunos estudios indican que el calcio y vitamina D complementarios pueden ser benéficos para los pacientes que están en tratamiento crónico con valproato.
Hematológicos: Trombocitopenia e inhibición de la fase secundaria de la agregación plaquetaria pueden reflejarse en alteración del tiempo de sangrado, petequias, contusiones, formación de hematomas, epistaxis y hemorragia franca (ver Precauciones generales e Interacciones medicamentosas y de otro género).
Linfocitosis relativa, macrocitosis, hipofibrinogenemia, leucopenia, eosinofilia, anemia incluyendo macrocítica con o sin deficiencia de folato, inhibición de médula ósea, pancitopenia, anemia aplásica, agranulocitosis y porfiria aguda intermitente.
Hepáticos: Las elevaciones menores de las transaminasas (por ejemplo, AST y ALT) y LDH son poco frecuente y al parecer se relacionan con la dosis. Ocasionalmente, los resultados de pruebas de laboratorio incluyen aumentos de bilirrubina y cambios anormales en otras pruebas de función hepática. Estos resultados pueden reflejar una hepatotoxicidad potencialmente seria (ver Precauciones generales).
Endocrinos: Menstruaciones irregulares, amenorrea secundaria, aumento de mamas, galactorrea e hinchazón de las glándulas parótidas. Pruebas de función tiroidea anormales (ver Precauciones generales). Existen reportes espontáneos poco frecuentes de enfermedad ovárica poliquística. No se ha establecido una relación de causa y efecto.
Pancreáticos: Pancreatitis aguda incluyendo muertes (ver Precauciones generales).
Metabólicos: Hiperamonemia (ver Precauciones generales), hiponatremia y secreción inadecuada de la hormona antidiurética (HAD).
Existen reportes poco comunes de síndrome de Fanconi principalmente en niños.
Se han reportado disminuciones en las concentraciones de carnitina, aunque la relevancia clínica no se conoce.
Se ha presentado hiperglucemia y se relacionó con la muerte de un paciente con hiperglucemia no cetósica preexistente.
Genitourinarios: Enuresis e infección de vías urinarias.
Sentidos especiales: Se ha reportado pérdida del oído, ya sea reversible o irreversible; sin embargo, no se ha establecido una relación de causa y efecto. También se ha reportado dolor de oído.
Otros: Reacción alérgica, anafilaxia, edema de extremidades, lupus eritematoso, dolor óseo, aumento de tos, neumonía, otitis media, bradicardia, vasculitis cutánea, fiebre e hipotermia.
Manía: Aunque el valproato sódico I.V. no ha sido evaluado en eficacia y seguridad en el tratamiento de episodios maniacos asociados con el trastorno bipolar, los siguientes eventos adversos fueron reportados en 1% o más de los pacientes en dos estudios clínicos controlados con placebo de tabletas de divalproato sódico.
Organismo en general: Dolor en cuello, rigidez de nuca.
Sistema cardiovascular: Hipotensión, hipotensión postural, vasodilatación.
Sistema digestivo: Incontinencia fecal, gastroenteritis, glositis.
Sistema musculoesquelético: Artrosis.
Sistema nervioso: Agitación, reacción catatónica, hipoquinesia, hiperreflexia, disquinesia, vértigo.
Piel y anexos: Furunculosis, rash maculopapular, seborrea.
Sentidos especiales: Conjuntivitis, ojos secos, dolor ocular.
Sistema urogenital: Disuria.
Migraña: Aunque el valproato sódico I.V. no ha sido evaluado en eficacia y seguridad en el tratamiento de profilaxis de migraña, los siguientes eventos adversos fueron reportados en 1% o más de los pacientes en dos estudios clínicos controlados con placebo de tabletas de divalproato sódico.
Organismo en general: Edema facial.
Sistema digestivo: Boca seca, estomatitis.
Sistema urogenital: Cistitis, metrorragia, hemorragia vaginal.
Trastornos generales y anomalías en el lugar de administración: Poco frecuentes: hipotermia, edema periférico no grave. Frecuencia no conocida: riesgo de necrosis tisular local con inyecciones repetidas.
Caja con 10 ampolletas de 4 mL e instructivo anexo.
Vía de administración: Intravenosa. Léase instructivo anexo.
Dosis:
Crisis parciales complejas (para adultos, niños de 10 años de edad y mayores):
Monoterapia (tratamiento inicial): Iniciar con 10 a 15 mg/kg/día, incrementando 5 a 10 mg/kg/semana para lograr la respuesta clínica óptima, la cual se obtiene, generalmente, con dosis por debajo de 60 mg/kg/día. Si no se obtiene la respuesta óptima, se deben medir los niveles plasmáticos para determinar si se encuentran dentro del rango aceptado (50 a 100 µg/mL). No se recomienda usar dosis de valproato por arriba de 60 mg/kg/día.
La probabilidad de trombocitopenia aumenta significativamente en concentraciones plasmáticas de valproato por arriba de 110 µg/mL en mujeres y de 135 µg/mL en hombres. Los beneficios de mejorar el control de las convulsiones con dosis más altas debieran sopesarse contra la posibilidad de mayor incidencia de reacciones adversas (ver Precauciones generales).
Conversión a monoterapia: Iniciar con 10 a 15 mg/kg/día, incrementando 5 a 10 mg/kg/semana para lograr la respuesta clínica óptima, la cual se obtiene, generalmente, con dosis por debajo de 60 mg/kg/día. Si no se obtiene la respuesta óptima, se deben medir los niveles plasmáticos para determinar si se encuentran dentro del rango aceptado (50 a 100 µg/mL). No hay recomendaciones respecto a la seguridad de usar el valproato a dosis > 60 mg/kg/día. Ordinariamente, se debe disminuir la dosificación del antiepiléptico concomitante (AEC) en 25% cada dos semanas. La reducción debe iniciarse al principiar el uso del divalproato de sodio; o retrasarse una o dos semanas si hay la sospecha de que con la reducción ocurran convulsiones. La rapidez y la duración de la retirada del AEC pueden ser altamente variables, y se debe monitorear estrechamente a los pacientes, durante este periodo, acerca del aumento en la frecuencia de las convulsiones.
Tratamiento adjunto: El valproato de sodio puede agregarse al régimen del paciente en 10 a 15 mg/kg/día, incrementando 5 a 10 mg/kg/semana para lograr la respuesta clínica óptima, la cual se obtiene, generalmente, con dosis por debajo de 60 mg/kg/día; si no se obtiene la respuesta óptima, se deben medir los niveles plasmáticos para determinar si se encuentran dentro del rango aceptado (50 a 100 µg/mL). No se recomienda usar dosis de valproato por arriba de 60 mg/kg/día.
Si la dosis diaria total excede 250 mg, se debe dar en dosis divididas.
En un estudio de tratamiento adjunto de convulsiones parciales complejas en pacientes que recibían ya sea carbamazepina o fenitoína, además del valproato de sodio, no se requirió ajustar la dosis de los dos primeros; sin embargo, ya que el valproato tiene interacciones con éstos y otros AEC, se recomienda determinar las concentraciones plasmáticas de AEC desde el inicio del tratamiento.
Crisis de ausencia simple y compleja: La dosis inicial de valproato de sodio I.V. que se recomienda es de 15 mg/kg/día, aumentando 5 a 10 mg/kg/día cada semana hasta controlar las convulsiones o hasta que los efectos adversos eviten su incremento. La dosis máxima recomendada es de 60 mg/kg/día. Si la dosis diaria total excede 250 mg, se debe dar en dosis divididas.
No se ha establecido una buena correlación entre la dosis diaria, las concentraciones séricas y el efecto terapéutico. Sin embargo, las concentraciones séricas terapéuticas en la mayoría de los pacientes con crisis de ausencia estarán en el rango de 50 a 100 µg/mL. Algunos pacientes se controlarán con concentraciones menores y, otros, a mayores (ver Farmacocinética y farmacodinamia).
Conforme aumenta la dosis de valproato de sodio, se pueden afectar las concentraciones de fenobarbital y fenitoína (vea Interacciones medicamentosas y de otro género).
Los otros antiepilépticos no deben suspenderse abruptamente en los pacientes en quienes el fármaco se administre para prevenir convulsiones mayores, debido a la fuerte posibilidad de precipitar status epilepticus, con hipoxia y riesgo para la vida.
Tratamiento de reemplazo: Cuando el cambio es de valproato oral, la dosis diaria total de valproato de sodio inyectable debe ser equivalente al producto oral (vea Farmacocinética y farmacodinamia) y debe ser administrado en infusión durante 60 minutos (pero no a más de 20 mg/min), con la misma frecuencia que los productos para vía oral, aunque puede ser necesario el monitoreo de las concentraciones plasmáticas y el ajuste de dosis. El monitoreo debe ser estrecho en aquellos pacientes que reciben dosis cercanas a la máxima recomendada (60 mg/kg/día), en particular los que no reciben fármacos inductores enzimáticos. Si la dosis total excede 250 mg, se deben dar en régimen dividido. Sin embargo, la equivalencia mostrada entre valproato de sodio inyectable y en formas orales (divalproato de sodio), en el estado estable, sólo se evaluó en un régimen de cada seis horas. Se desconoce qué sucede si la inyección de valproato de sodio se da con menor frecuencia (2 a 3 veces al día), con niveles por debajo de los que resultan de la dosis oral dada en el mismo régimen; por tanto, si se administra valproato de sodio inyectable 2 o 3 veces al día, se requerirá un monitoreo estrecho de los niveles plasmáticos.
Recomendaciones generales de dosificación:
Administración en pacientes de edad avanzada: Debido a una disminución en la depuración de valproato no unido y posiblemente a una mayor sensibilidad a la somnolencia en los ancianos, la dosis inicial debe reducirse en estos pacientes. La dosis debe aumentarse más lentamente y con un monitoreo periódico de ingesta de líquidos y alimentos, deshidratación, somnolencia y otros eventos adversos. En pacientes con menor ingesta de alimentos o líquidos y en pacientes con somnolencia excesiva debe considerarse reducir o suspender la dosis de valproato.
La dosis terapéutica máxima debe lograrse con base en la tolerabilidad y respuesta clínica (ver Precauciones generales).
Adolescentes y varones en edad fértil
Se recomienda que el tratamiento con VALPROATO SÓDICO lo inicie y supervise un especialista con experiencia en el tratamiento de la epilepsia (ver PRECAUCIONES GENERALES RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA).
Modo de empleo:
En el caso de reemplazo simple (por ejemplo, en el caso de cirugía planeada): entre 4 y 6 horas después de la última dosis oral, administre valproato de sodio por vía intravenosa en una solución de cloruro de sodio al 0.9% para inyección,
- Ya sea por infusión continua durante 24 horas
- O en dosis fraccionadas en 4 infusiones de una hora por día, en la dosis previa.
En casos que requieran alcanzar una concentración efectiva de plasma rápidamente y luego mantenerla:
Se proporciona una inyección intravenosa de una dosis en bolo de 15 mg/kg durante 5 minutos antes de la infusión continua con un flujo de 1 mg/kg/hora que se ajustara progresivamente para alcanzar los niveles de ácido valproico en la sangre de 75 mg/L. Luego ajuste el flujo de acuerdo con al curso de la condición clínica.
En cuanto se detenga la infusión, reanudar la terapia oral hará posible asegurar la compensación inmediata de las cantidades eliminadas, se dará ya sea en la dosis previa o después de un ajuste de la dosis.
Después de la dilución en cloruro de sodio al 0.9%, se ha demostrado la estabilidad física y química de la solución diluida durante 24 horas. Sin embargo, desde una perspectiva microbiológica, se debe usar el medicamento de inmediato.
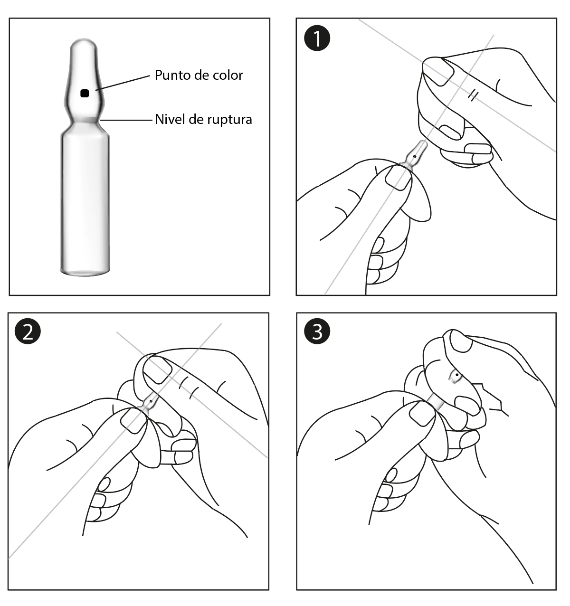
Instrucciones para abrir las ampolletas:
La ampolleta presenta un área frágil en el cuello, marcado por un punto de color (único punto de abertura). Para abrir la ampolleta de forma correcta, es esencial aplicar presión en esta área conforme al siguiente procedimiento:
- Con una mano, sostenga el cuerpo de la ampolleta firmemente mientras la cabeza de la ampolleta está extendida, con el punto de color hacia usted.
- Con la otra mano, tome la parte superior de la ampolleta, con el dedo índice detrás del cuello de la ampolleta y el pulgar en el punto de color como se muestra en el diagrama (ambos pulgares están perpendiculares).
- Mientras sostiene cada parte de la ampolleta firmemente, rómpala con un chasquido al presionar sobre ella.
Eventos adversos relacionados con la dosis: La frecuencia de efectos adversos (en particular elevación de enzimas hepáticas y trombocitopenia) puede estar relacionada con la dosis. La probabilidad de trombocitopenia al parecer aumenta significativamente a concentraciones totales de valproato de ≥ 110 µg/mL (mujeres) o ≥ 135 µg/mL (varones) (ver Precauciones generales). Se debe valorar el beneficio de un mejor efecto terapéutico con dosis más altas contra la posibilidad de una mayor evidencia de reacciones adversas.